

DeepMind - Презентация AlphaFold в EBI
February 07, 2019 4 min read
Два месяца назад весь мир облетела новость, что DeepMind выиграл известное соревнование по предсказанию 3D-структур белков CASP, порвав всех биоинформатиков с впечатляющим отрывом. Многие люди из мира биотеха теперь пытаются осознать, 'что это было'? Революция или эволюция, наука или инженерия, талант или финансирование? Волею судеб я когда-то оказался совсем недалеко от этой области науки, поэтому потратил несколько дней чтобы разобраться в деталях - а между тем в EBI приехал наводить мосты ведущий инженер проекта Эндрю Сеньор из DeepMind.

Что вообще произошло?
В этом посте я сначала расскажу, что такое CASP и зачем его проводят, потом постараюсь быстро обрисовать как решали эту задачу раньше, и за счет чего победил DeepMind. А потом поговорим о том, где заканчивается наука и начинается инженерия, и как ими заниматься в академии и индустрии.
Какую проблему решал DeepMind?
Основную работу в наших клетках осуществляют белки. Белки состоят из последовательности аминокислот (например, "MAKFGEWTTPFTNS" - всего бывает 20 разных аминокислот, каждой соответствует своя буква вроде "M" - метионин). Прочитать последовательность белка очень дешево, но расшифровать 3D-структуру белка (как та, что на обложке этой статьи) крайне дорого и трудоемко. Но именно 3D-структура содержит ценную информацию для синтеза новых лекарств - например, по ней можно подобрать вещества, которые связываются с этим белком и подавляют его функцию - такие вещества могут стать новыми лекарствами.
Хорошо, а как насчет всяких законов Ньютона и т.п.: если мы знаем последовательность белка, может быть мы сможем предсказать, как свернется белок в пространстве просто под действием сил? Не получается. Куча бывших физиков и математиков, сбежавшихся в биоинформатику, десятилетиями бились с этой задачей практически безрезультатно. Около 25 лет назад был создан конкурс CASP - Critical Assessment of Protein Structures - где участникам предлагалось предсказать 3D-структуру белка по последовательности.
После 10 лет стагнации, на CASP'10 в 2012 был достигнут большой прогресс. Известно было, что у одного и того же белка в разных видах меняется последовательность (например, пока человек происходил из обезьяны, пара аминокислот в альбмуине у него изменилась). Однако пространственная структура белков даже при изменении последоавтельности меняется довольно слабо. Тогда этот факт можно использовать: люди додумались смотреть на коэволюцию аминокислот. Например, если в человеческой версии белка в одном месте последовательности была положительно заряженная аминокислота, а в другом - отрицательно, а у аналогичного белка из обезьяны - наоборот, то можно предположить, что эти аминокислоты неспроста так коэволюционируют - видимо, они взаимодействуют, а значит, сближены в пространстве.
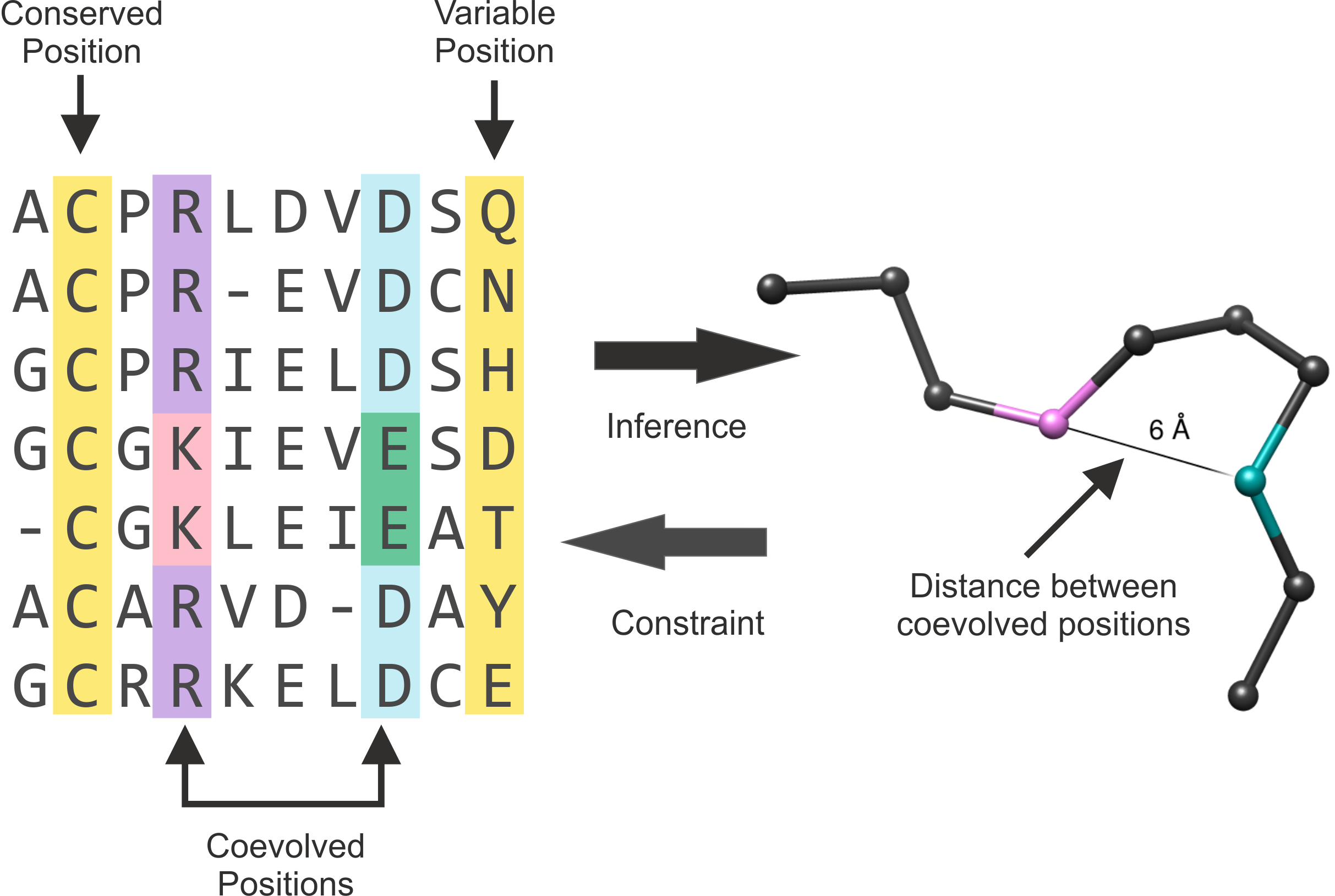
Записывая однотипные белки друг под другом, биоинформатики стали строить так называемые выравнивания, и выводить из них структуры белков.
Спустя несколько лет появилась идея, где можно достать гораздо больше данных для вычисления коэволюции: появилась метагеномика. Дело в том, что большинство организмов, последовательности генов которых нам известны, хорошо культивируются в лабораторных условиях. Но, к примеру, среди бактерий основную массу культивировать не выходит.
Поэтому вместо того, чтобы пытаться выделить одну бактерию и отсеквенировать именно ее, народ стал просто "зачерпывать" ДНК из окружающей среды и секвенировать (прочитанная из среды ДНК стала называться метагеномной) - и в этом бульоне оказалось гораздо больше разнообразия белков, чем было известно до этого. Сергей Овчинников из Гарварда придумал, что это разнообразие можно использовать и для получения данных о коэволюции - и предсказания удалось улучшить еще больше.
Затем в районе 2016 - CASP'12 - случилась новая революция: все стали использовать нейронки для оптимизации структур. Со временем устоялся более-менее один и тот же подход.
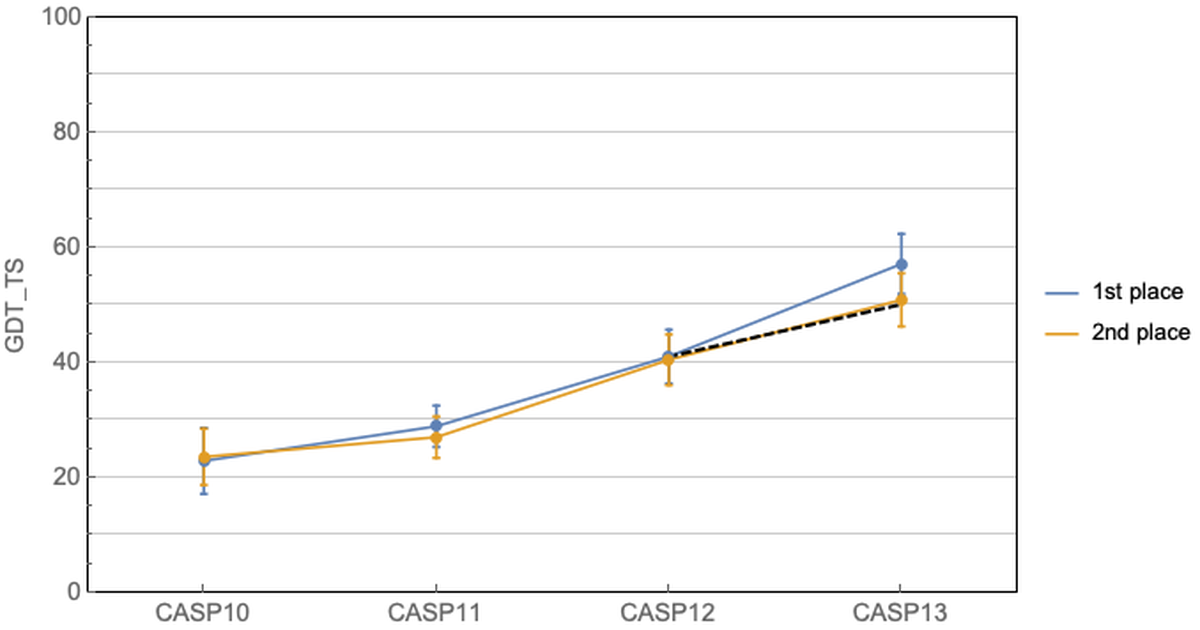
К началу CASP'13 несколько научных групп сообразили, что про каждую пару остатков нужно говорить не просто сближены они или нет, а стараться определить расстояние. И тут бы одна из групп-пионеров в этой области и победила... Но на сцену ворвался DeepMind.
Как все было?
Все началось как хакатон. Один из сотрудников DeepMind для интереса попробовал за выходные запустить предсказание 3D-структуры белка, и добился неплохих результатов.
Затем сотрудники DeepMind пообщались со структурным биоинформатиком и соавтором системы классификации пространственных структур CATH Дэвидом Джонсом из Crick Institute при University College London (там в свое время переучивался на машиниста Демис Хассабис). Тот объяснил им, как устроен современный биоинформатический пайплайн предсказания 3D-структур белков.
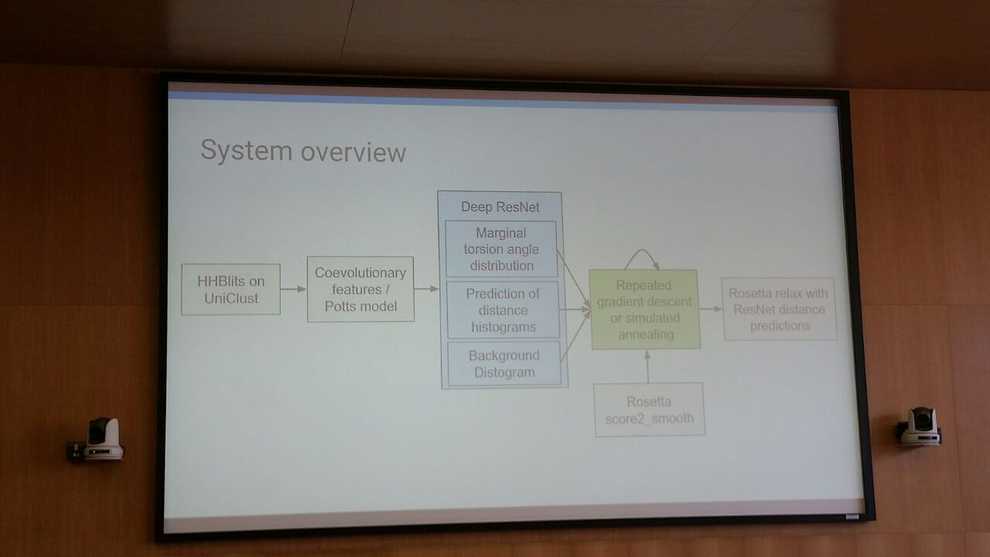
Ребята из DeepMind не стали сильно заморачиваться с биологией, и сразу взялись за серединку этого пайплайна - нейронную сетку - и инженерию фич для нее.
Я не смогу пересказать всего, что сделала эта команда. Они перебрали около сотни разных структур нейронок и остановились вот на такой:

В качестве обучающей выборки они взяли все извесные структуры из главной мировой бызы 3D-структр PDB, которая содержит в себе около 150 тысяч структур, но большинство из них сильно повторяются, так что реальный размер выборки составлял лишь порядка 30 тысяч структур. Это очень мало для машинного обучения.

Поэтому они сделали изящную аугментацию данных, "пошатав" отдельные спирали и листы известных 3D-структур, что, кстати, вполне соответствует биологической природе белков - они так в растворе и шатаются.
Они придумали также огромное количество других приемчиков. Они хитрили с определением границ доменов. Они заменили матрицу расстояний между остатками на матрицу распределений расстояний. Они посреди конкурса поменяли градиент с симулированного отжига на l-BFGS. Они колдовали с торсионными углами в белках, чтобы сделать их реалистичными...
И что дальше?
DeepMind готовит по работе публикацию. Они довольно свободно и подробно рассказывают о том, что было сделано, но не планируют пока публиковать исходный код. По слухам это объясняется тем, что они еще не все выжали из выбранного подхода и могут сделать еще лучше. Кроме того, они ничего не понимают в биологии, а после всех неимоверных усилий, что были затрачены, кажется, обидно не получить за них какое-то вознаграждение. В EBI они по этому поводу усиленно наводили мосты и интересовались, к кому бы теперь с этим молотком пойти за гвоздями.
Так все же что это было?
Это была абсолютно титаническая инженерная работа. Они практически не коснулись биологии совсем, почти ничего нового не придумали. Просто очень, очень хорошо исполнили machine learning'овую часть и положили на нее очень много усилий превосходных инженеров. По-видимому, к чему-то подобному шла и биоинформатика, и наверное к следующему-позаследующему CASP'у пришла бы и независимо. Победа DeepMind - это не революция, а эволюция и преимущественно не фундаментальная наука, а виртуозная инженерия.
В соавторах работы 15+ человек, по-видимому, основной вклад внесли шестеро. Сам Эндрю Сеньор занимался нейронками еще с 1994 года - представьте, какой колоссальный опыт был накоплен им за четверть века.
Очень советую также прочитать вот этот блестящий блог пост - там один из специалистов в данной области очень понятным языком рассказывает обо всем случившемся, я много информации почерпнул именнно у него.

Written by Boris Burkov who lives in Moscow, Russia, loves to take part in development of cutting-edge technologies, reflects on how the world works and admires the giants of the past. You can follow me in Telegram